4. Settings for RASPA¶
The RASPA package can perform MC and MD calculations, for a variety of systems.
In this tutorial we use this program to perform Grand Canonical (GCMC) calculations in nanoporous structures. Here it is basic input:
SimulationType MonteCarlo
NumberOfInitializationCycles ***
NumberOfCycles ***
PrintEvery ***
RestartFile no
RestartStyle Raspa
CutOff ***
Forcefield UFF-TraPPE
ChargeMethod ***
Framework 0
FrameworkName ***
UnitCells * * *
ExternalTemperature ***
ExternalPressure ***
Component 0 MoleculeName methane
MoleculeDefinition TraPPE
BlockPockets yes/no
BlockPocketsFileName *****
TranslationProbability ***
RotationProbability ***
ReinsertionProbability ***
SwapProbability ***
CreateNumberOfMolecules ***
These settings are imported in AiiDA as a ParameterData object, with the “shape” of a python dictionary.
You should fill the input settings, replacing asterisks with meaningful values.
Consider that the forcefield parameters that you will be using to model the framework-molecule interactions are on your remote computer:
$RASPA_DIR/share/raspa/forcefield/UFF-TraPPE/contains the Lennard-Jones parameters for the framework atoms, from UFF, and for methane, from TraPPE$RASPA_DIR/share/raspa/molecules/TraPPE/contains the filemethane.defwith the critical T and P and acentric factor of methane (why?) and the geometry of this gas molecule. Note that we are using a forcefield where methane is modelled as a single point!
As an extra exercise you may decide to use another popular forcefield
for the framework atoms:
DREIDING. Note
that, since it does not contain all the atoms of the periodic table, it
is common practice to use UFF parameters for the missing ones.\ You can
find this forcefield as
$RASPA_DIR/share/raspa/forcefield/DREIDING-TraPPE/, and remember to
modify Forcefield DREIDING-TraPPE in the input.
You can even modify, create or import your own forcefield: just create
the folder $RASPA_DIR/share/raspa/forcefield/{ffname}/, copy the
file formatting of the other forcefield .def files and specify
Forcefield {ffname} in the input.
4.1. A few hints for the settings:¶
BlockPocketsandBlockPocketsFileNamewill be filled by AiiDA: if Zeo++ finds some non accessible pore volume, it can generate a .block file with the positions and the radii of blocking spheres. These spheres are inserted in the framework to prevent Raspa from inserting molecules in the non accessible pore.You need to choose
NumberOfInitializationCyclesandNumberOfCycles. Consider that for every cycle every molecule of your system is selected to perform a MC movement that may be accepted or rejected. If your system is empty or there are a few particles, Raspa tries to perform GCMC insertions (“swaps”) to fill your system (see UMS, Algorithm 13). The number of particles continues to fluctuate according to the Grand Canonical ensemble, and consider that afterNumberOfInitializationCyclescycles, Raspa start to compute the average number of particles forNumberOfCyclescycles. Therefore, the number of cycles should be large enough to compute statistics correctly but not too large to waist computational times after your uncertainty is already low. Standard deviation is computed using Block Averages (see UMS pag. 529).
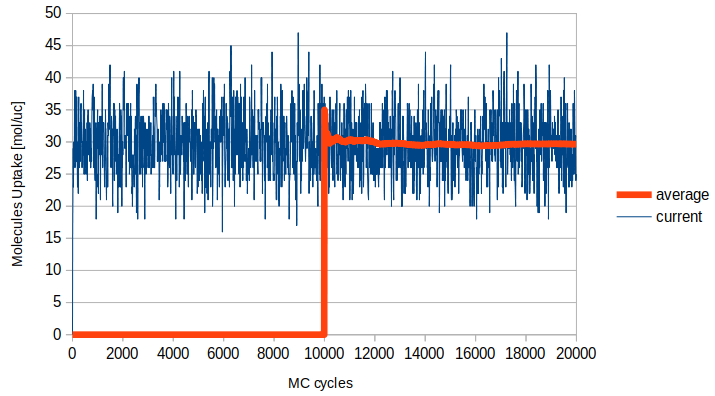
Fig. 4.3 Here an example of the value fluctuation and average, with the averaging starting after 10k cycles.¶
You have to chose the probability for the different swap movements. The total will be normalized: for example if you assign 2.0, 1.0, 1.0, 1.0, this will be normalized as 40%, 20%, 20%, 20% probability for each move.
TranslationProbability: rigid displacement by a random distance. The maximum displacement is scaled during the simulation to achieve an acceptance ratio of 50%.RotationProbability: rotation around its starting beadReinsertionProbability: reinsertion of the particle in a random position of the unit cell.SwapProbability: insertion or deletion move. Whether to insert or delete is decided randomly with a probability of 50% for each. The swap move imposes a chemical equilibrium between the system and an imaginary particle reservoir for the current component.
Choose a reasonable
cutoff(Angstrom) to exclude negligible Lennard-Jones interaction between far particles. Consider that the higher the cutoff the more you will have to expand your structures!You can choose
ChargeMethodtoNoneorEwald, depending if you want or not to compute Coulombic interactions.FrameworkNamewill be filled by AiiDAUnitCellswill be filled by the multiply_unit_cell(cif) function: it gives the cell expansion that you need to perform according to the cutoff you chose.With
CreateNumberOfMoleculesyou can specify the number of particles that will be inserted (randomly) in your system at the start of your simulation.