Running external codes
Contents
Running external codes¶
In this section you will learn how to run external codes with AiiDA through a calculation plugin.
We’ll be using the Quantum ESPRESSO package to launch a simple density functional theory (DFT) calculation and check its results. More specifically, we’ll be performing a self-consistent field (SCF) electronic relaxation for crystalline silicon using the PBE exchange-correlation functional.
The typical way one would do this without AiiDA is by writing an input file and feeding it to the pw.x code of the Quantum ESPRESSO package.
This input would contain the information of the crystal structure (atomic coordinates, cell vectors) as well as any other parameters that need to be specified (type of calculation, energy cutoffs, k-point grid, etc).
A typical Quantum ESPRESSO input file looks something like this:
&CONTROL
calculation = 'scf'
outdir = './out/'
prefix = 'test_run'
pseudo_dir = './pseudo/'
/
&SYSTEM
ecutrho = 2.4000000000d+02
ecutwfc = 3.0000000000d+01
ibrav = 0
nat = 2
ntyp = 1
/
&ELECTRONS
conv_thr = 8.0000000000d-6
electron_maxstep = 80
mixing_beta = 4.0000000000d-01
/
ATOMIC_SPECIES
Si 28.085 Si.pbe-n-rrkjus_psl.1.0.0.UPF
ATOMIC_POSITIONS angstrom
Si 5.8004619750 3.3488982827 2.3680286852
Si 3.8669746500 2.2325988551 1.5786857901
K_POINTS automatic
4 4 4 0 0 0
CELL_PARAMETERS angstrom
3.8669746500 0.0000000000 0.0000000000
1.9334873250 3.3488982827 0.0000000000
1.9334873250 1.1162994276 3.1573715803
One would also need to provide the pseudopotentials for all atomic species present (in this case, just Si) inside the pseudo_dir (in this case, the pseudo folder inside the same directory).
When all of this is in place, the only thing left would be to run the code.
If you are not very familiar with Quantum ESPRESSO, it might be helpful to first manually run this example by yourself.
You can find the instructions in the dropdown below!
Try it yourself!
Create a new test_run folder and copy the inputs shown above into the qe.in file.
Next, make a pseudo subfolder and download the pseudopotential for silicon there:
$ wget https://aiida-tutorials.readthedocs.io/en/tutorial-2021-intro/_downloads/ea4486c5b943d172e522da9bc9dc56ae/Si.pbe-n-rrkjus_psl.1.0.0.UPF
The final directory structure should look something like this:
test_run/
├── pseudo/
│ └── Si.pbe-n-rrkjus_psl.1.0.0.UPF
└── qe.in
Now you can just run the code:
$ pw.x < qe.in | tee -a qe.out
By using the tee command, the Quantum ESPRESSO output is automatically printed to both the stdout and qe.out file.
Once the calculation is complete (you will see JOB DONE. at the end), you can check the outputs generated: mainly the qe.out file, but there might also be some interesting data in the files inside of the out subfolder.
Running with AiiDA¶
![]() AiiDA plugin packages
AiiDA plugin packages
Besides aiida-quantumespresso, there are many other AiiDA plugin packages available for simulation codes, all of which are listed in the AiiDA plugin registry.
To run Quantum ESPRESSO through AiiDA you will make use of the aiida-quantumespresso package, which contains everything needed for both codes to be able to interact with one another. You will not create the input file yourself, but provide AiiDA with the input nodes that contain the information that you want to use and then just instruct it to run them. AiiDA will then create the input file, run the calculation, and extract all relevant data from the output files into output nodes automatically.
Fig. 5 shows the resulting provenance graph associated with the procedure described above applied to our original example. This one was drawn by hand, but you will later generate one using AiiDA tools.
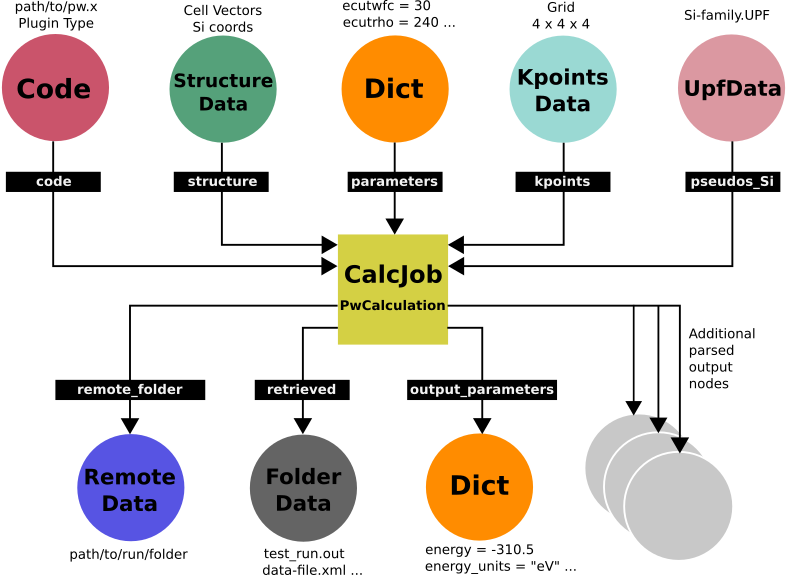
Fig. 5 Graph representing the provenance for a typical Quantum ESPRESSO calculation.¶
You can see the calculation node represented by a square, while all the data nodes are represented by circles.
The input data nodes are the ones above the calculation node, with arrows pointing towards it, while the output data nodes are the ones below, and have the arrows coming from the calculation node.
Symbolized in gray are additional optional output nodes that might be present or not depending on the type of calculation performed (e.g., an output StructureData if you performed a relaxation calculation, a TrajectoryData for molecular dynamics, etc.).
You can also see in a black box the label for all the links that connect the calculation with the data.
You will notice that the information that goes into the test_run.in input file can be dispersed into different data nodes, such as:
the
StructureDatanode with link labelstructurenode contains the data for the blocks ATOMIC_POSITIONS and the CELL_PARAMETERS.the
KpointsDatanode with link labelkpointscontains the data for the K_POINTS block.the
UpfDatanode with link labelpseudos_Sicontains the data for the ATOMIC_SPECIES block (including the pseudopotential file to be copied in thepseudo_dir).the
Dictnode with link labelparameterscontains the rest of the data (for the &CONTROL and &SYSTEM namelists).
Once the inputs files are prepared from these nodes, they are copied into the computer or cluster where the calculation will run.
AiiDA immediately generates a RemoteData node as part of this submission procedure; this node can be thought as a symbolic link to the remote folder where the files are copied.
The other output nodes are created once the calculation has finished, after the retrieval and parsing steps.
The retrieved node contains the relevant raw output files copied back and stored into the AiiDA repository; all other output nodes are added by the parser and contain information taken from those files.
In the following sections you will first deal with setting up the inputs: you will learn to create, import, load, etc. the data nodes themselves, and then you will see how to connect them to the calculation before launching it. Afterwards you will see how to submit the calculation to the engine, follow its advancement, and analyze the resulting output nodes.
Codes and plugins¶
You may have noticed that we did not mention anything about the Code input when describing Fig. 5 in the previous section.
Indeed, before we go further, it is important to take a minute to point out and understand the distinction between several related entities involved in the launching of a calculation.
These entities are:
The actual code (
pw.xfrom Quantum ESPRESSO)The calculation plugin (
quantumespresso.pwfrom aiida-quantumespresso)The code node
The calculation node
The code (1) is the program that knows how to perform the procedure one wants to apply (in this case, the SCF DFT). This can be used with or without AiiDA, and needs to already be installed and configured independently from your AiiDA environment.
The calculation plugin (2) contains the instructions that indicate to AiiDA how a code (1) works in general.
This includes information about what the input files look like, how to generate them from data (input) nodes, what outputs are produced, and how to parse them into data (output) nodes.
This is what you get when you pip install one of AiiDA’s plugin packages.
The code node (3) is a data node that contains the instructions for AiiDA to execute a specific instalation of the code (1). It does have a reference to which plugin (2) it needs to use, but on top of that it also includes: the path to the installed executable, any environment variable required to run it, etc. As this is a data node, it then becomes part of the provenance of every process that uses it (4).
Finally, a calculation node (4) stands for a specific execution of the code (1). It uses the general information of the code plugin (2) when it is being setup, and the specifics of its input code node (3) for running the actual code.
Important
Both the Quantum Mobile virtual machine and the AiiDAlab cluster come pre-configured with all that you need related to codes. That is, they already include:
The Quantum ESPRESSO code with
pw.xThe
aiida-quantumespressoplugin package withquantumespresso.pwA pre-configured code node ready to use the local
pw.xcode
You can check this by running the following:
$ verdi code list
# List of configured codes:
# (use 'verdi code show CODEID' to see the details)
* pk 192 - pw@localhost
* pk 193 - projwfc@localhost
* pk 194 - dos@localhost
Make a note of the PK or label of the pw code (in the case above, pw@localhost) since you’ll need to replace it in code snippets later in this tutorial.
Setting up a new pw code
If by any chance you do not have a pw code node already set up in your environment or you need to set up a new one, you can do so with the following commands:
$ verdi code setup --label pw --computer localhost --remote-abs-path /opt/conda/bin/pw.x --input-plugin quantumespresso.pw --non-interactive
Success: Code<2> pw@localhost created
You now should be able to see this new code when you execute verdi code list.
Structure and pseudopotentials¶
Besides the code, there are other two nodes of the ones shown in on Fig. 5 that require special consideration: the structure and the pseudos.
This is because they contain important physical information and it is hard to create them from scratch.
We will first see how to easily import a structure from a file into the AiiDA database.
Download the 
Si.cif structure file in your work environment.
You can open this file and check it out with your editor of choice as it has the information in a human-readable format.
Tip
You can download any file directly into the AiiDAlab JupyterHub cluster using wget.
All you need to do is copy the link for the download (for example, right clicking on the link above and selecting “Copy link”) and then just use wget:
$ wget https://aiida-tutorials.readthedocs.io/en/tutorial-2021-intro/_downloads/92e2828a59fc133b391bbf62f0fd1b59/Si.cif
Now import it into your database with the verdi CLI.
$ verdi data structure import ase Si.cif
Successfully imported structure Si2 (PK = 1)
The output of verdi data structure import shows the PK of the structure node you just created (your value may be different from the one shown here).
Make a note of this PK, as you will need to replace it in code snippets later in this tutorial.
Should you forget it, you can always have a look at the structures in the database using:
$ verdi data structure list
Id Label Formula
---- ------- ---------
1 Si2
For managing pseudopotentials, there is another plugin package installed in the default environment: aiida-pseudo.
This package comes with its own CLI (that uses aiida-pseudo <command> instead of verdi <command>) to interact with its features.
To get a list of all available pseudopotentials, simply run:
$ aiida-pseudo list
Label Type string Count
----------------------------------- ------------------------- -------
SSSP/1.1/PBEsol/precision pseudo.family.sssp 85
SSSP/1.1/PBEsol/efficiency pseudo.family.sssp 85
SSSP/1.1/PBE/precision pseudo.family.sssp 85
SSSP/1.1/PBE/efficiency pseudo.family.sssp 85
You can see above that the AiiDAlab cluster already comes with
Note
If you are using the Quantum Mobile virtual machine, you will need to install the SSSP pseudopotentials.
Luckily, doing it with aiida-pseudo is easy!
All you need to do is run:
$ aiida-pseudo install sssp
Info: downloading selected pseudo potentials archive... [OK]
Info: downloading selected pseudo potentials metadata... [OK]
Info: unpacking archive and parsing pseudos... [OK]
Success: installed `SSSP/1.1/PBE/efficiency` containing 85 pseudo potentials
The output already indicates the process was successful and shows you the label for the new family group.
You can always run aiida-pseudo list if you want to double check or if you later forget the ID.
Exercise¶
Follow the previous procedure to install the pseudo-dojo family; you will need it for other exercises!
Remember that you can run aiida-pseudo install -h to check the exact command needed for the family you want.
Since we need the pseudopotentials to be in the UPF format, be sure to also run the help command once you know the family name to find the option required for this (aiida-pseudo install <FAMILY_NAME> -h).
Solution
$ aiida-pseudo install pseudo-dojo -f upf
Info: downloading selected pseudopotentials archive... [OK]
Info: downloading selected pseudopotentials metadata archive... [OK]
Info: unpacking archive and parsing pseudos... [OK]
Info: unpacking metadata archive and parsing metadata... [OK]
Success: installed `PseudoDojo/0.4/PBE/SR/standard/upf` containing 72 pseudopotentials
Preparing the calculation¶
The setup for preparing a calculation must be done through the Python ORM, so let’s start the verdi shell:
$ verdi shell
There are several ways to setup and launch processes with AiiDA. We will now show you how to do it using a builder, which is a tool that is particullarly convenient when manually preparing the inputs. The simplest way to get a builder for a calculation is from a code node, so load the one we checked at the begining of this module:
In [1]: code = load_code(<CODE_PK>)
Now to get a builder for the calculation all you have to do is use the get_builder() method of the code node:
In [2]: builder = code.get_builder()
All possible inputs for the process are properties of the builder object called ports.
You can set them up and access them by using the builder.<input_name> syntax.
Moreover, when you use the get_builder() method to get a calculation builder, it already comes with the code node in the adequate input port:
In [3]: builder.code
Out[3]: <Code: Remote code 'qe-pw-6.5' on localhost, pk: 7, uuid: 72655d43-5b17-4547-be38-0338773eaced>
One nifty feature of the builder is the ability to use tab completion.
Try it out by typing builder. + <TAB> in the verdi shell (you can navigate the options with the keyboard arrows).
You can also get more information on an input by adding a question mark ? and hitting enter.
For example:
In [4]: builder.structure?
Type: property
String form: <property object at 0x7f3393e81050>
Docstring: {"name": "structure", "required": "True", "valid_type": "<class 'aiida.orm.nodes.data.structure.StructureData'>", "help": "The input structure.", "non_db": "False"}
In the output Docstring you can see that the structure input is required (as opposed to being optional) and needs to be of node type StructureData.
Unlike what happened with the code input, the rest of the ports (like the structure) don’t come with anything pre-loaded on them.
You can check this by, for example, executing builder.structure and getting an empty output.
Input selection¶
Let’s supply the builder with the structure we just imported.
In [3]: structure = load_node(<STRUCTURE_PK>)
...: builder.structure = structure
Next, for the pseudos input, you will need a dictionary that maps the elements (Si) to the pseudopotentials you want to use.
Let’s first load the pseudopotential family we installed before with aiida-pseudo:
In [5]: pseudo_family = load_group('SSSP/1.1/PBE/efficiency')
The selection of pseudos for any structure can be easily performed using the get_pseudos() method of the pseudo_family:
In [6]: pseudos = pseudo_family.get_pseudos(structure=structure)
If we now check the contents of the pseudos variable:
In [6]: pseudos
Out[6]: {'Si': <UpfData: uuid: afa12680-efd3-4e9a-b4a7-b7a69ee2da51 (pk: 69)>}
We can see that it is a simple dictionary that maps the 'Si' element to a UpfData node, which contains the pseudopotential for silicon in the database.
Let’s pass the pseudos to the builder:
In [7]: builder.pseudos = pseudos
Of course, we also have to set the computational parameters of the calculation. You will first set up a dictionary with a simple set of input keywords for Quantum ESPRESSO:
In [8]: parameters = {
...: 'CONTROL': {
...: 'calculation': 'scf', # self-consistent field
...: },
...: 'SYSTEM': {
...: 'ecutwfc': 30., # wave function cutoff in Ry
...: 'ecutrho': 240., # density cutoff in Ry
...: },
...: }
The builder.parameters port requires a Dict node (you can verify this by running builder.parameters?), so this previous content must be converted to one before being passed to the builder:
In [9]: builder.parameters = Dict(dict=parameters)
Note
The node assigned to builder.parameters does not necessarily need to be stored in the database.
Whenever you launch the calculation, AiiDA will take care of storing any unstored input node.
The k-points mesh can be supplied via a KpointsData node.
To create it, you will first load the corresponding class using the DataFactory, a useful and robust tool which recognizes data types based on their entry point (e.g. 'array.kpoints' in this case):
In [10]: KpointsData = DataFactory('array.kpoints')
Once the class is loaded, defining the k-points mesh and passing it to the builder is easy:
In [11]: kpoints = KpointsData()
...: kpoints.set_kpoints_mesh([4,4,4])
...: builder.kpoints = kpoints
Finally, we can also specify the resources we want to use for our calculation from the computer or cluster where we will be running. These details are stored in a sub-port of the metadata:
In [12]: builder.metadata.options.resources = {'num_machines': 1}
The resources port (or more generally, the whole metadata port) does not expect a Node but a regular Python Dict.
Indeed, not all inputs for a calculation will require AiiDA Nodes.
Submitting the calculation¶
Great, you are all set! Now all that is left to do is to submit the builder to the daemon.
In [13]: from aiida.engine import submit
...: calcjob_node = submit(builder)
Let’s exit the verdi shell using the exit() command and have a look how your calculation is doing.
You can check the list of processes stored in your database using verdi process list:
$ verdi process list
PK Created Process label Process State Process status
---- --------- --------------- --------------- ----------------
90 6s ago PwCalculation ⏹ Waiting Monitoring scheduler: job state RUNNING
Total results: 1
Info: last time an entry changed state: 6s ago (at 23:14:25 on 2021-02-09)
We can see above the PwCalculation you have just set up, i.e. the process that runs a Quantum ESPRESSO pw.x calculation, which is shown to be in the Waiting state.
Note
If the state of the calculation is Created instead, it may be that the daemon is not running.
In this case you should also see a warning at the end of the output:
$ verdi process list
PK Created Process label Process State Process status
---- --------- --------------- --------------- ----------------
90 36s ago PwCalculation ⏹ Created
Total results: 1
Info: last time an entry changed state: 36s ago (at 23:14:25 on 2021-02-09)
Warning: the daemon is not running
If this is the case, you need to start the daemon using:
$ verdi daemon start
Starting the daemon... RUNNING
If you see an empty list, you calculation may already be over (it should take less than one minute to complete) and so it won’t show by default in the verdi process list.
To see all processes, use the -a, --all option:
$ verdi process list -a
PK Created Process label Process State Process status
---- --------- --------------- --------------- ----------------
90 8m ago PwCalculation ⏹ Finished [0]
Total results: 1
Info: last time an entry changed state: 22s ago (at 23:22:07 on 2021-02-09)
Exploring the results¶
Use the PK of the PwCalculation to get more information on it:
$ verdi process show <PK>
This will produce a lot of details on the calculation and its in- and outputs:
Property Value
----------- ------------------------------------
type PwCalculation
state Finished [0]
pk 90
uuid 85e38ed3-bb42-4a4b-bd28-d8031736193e
label
description
ctime 2021-02-09 23:14:24.899458+00:00
mtime 2021-02-09 23:22:07.100611+00:00
computer [1] localhost
Inputs PK Type
---------- ---- -------------
pseudos
Si 69 UpfData
code 2 Code
kpoints 89 KpointsData
parameters 88 Dict
structure 1 StructureData
Outputs PK Type
----------------- ---- --------------
output_band 93 BandsData
output_parameters 95 Dict
output_trajectory 94 TrajectoryData
remote_folder 91 RemoteData
retrieved 92 FolderData
As you can see, AiiDA has tracked all the inputs provided to the calculation, allowing you (or anyone else) to reproduce it later on.
AiiDA’s record of a calculation is best displayed in the form of a provenance graph. You can generate one by running the following verdi command using the PK of your calculation node:
$ verdi node graph generate <PK>
The command will write the provenance graph to a file named <PK>.dot.pdf.
It should look like this:
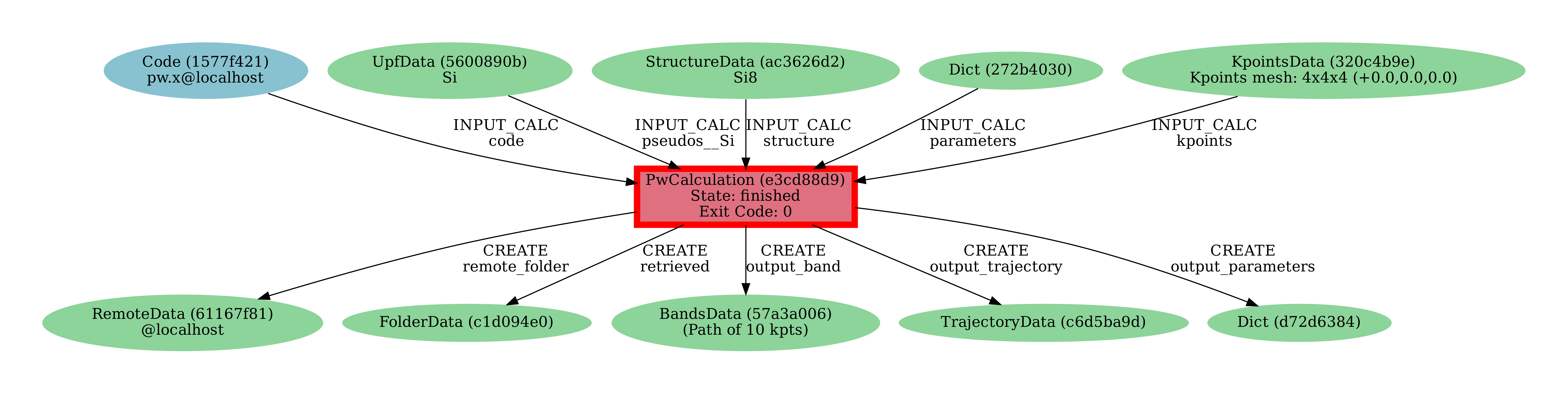
Fig. 6 Provenance graph for a single Quantum ESPRESSO calculation.¶
Let’s have a look at one of the outputs, i.e. the output_parameters Dict node.
Grab the PK (95 in the example above) and load the node in the verdi shell:
In [1]: node = load_node(<PK>)
You can get the contents of this Dict node using the get_dict() method:
In [2]: d = node.get_dict()
There is a lot of information stored in the output parameters dictionary, parsed from the Quantum ESPRESSO output files.
A basic result is the system energy:
In [3]: d['energy']
Out[3]: -310.56907438957
Moreover, you can also easily access the input and output files of the calculation using the verdi CLI:
$ verdi calcjob inputls <PK> # Shows the list of input files
$ verdi calcjob inputcat <PK> # Shows the input file of the calculation
$ verdi calcjob outputls <PK> # Shows the list of output files
$ verdi calcjob outputcat <PK> # Shows the output file of the calculation
$ verdi calcjob res <PK> # Shows the parser results of the calculation
Exercises¶
(1) A few questions you could answer using these commands:
How many atoms did the structure contain? How many electrons?
How many k-points were specified? How many k-points were actually computed? Why?
How many SCF iterations were needed for convergence?
How long did Quantum ESPRESSO actually run (wall time)?
(2) Now launch another pw calculation, except this time instead of using the SSSP pseudopotentials you can use the pseudo-dojo.
Is there any difference in the energy, number of iterations, wall time, etc?
Important
Key takeaways
The inputs of calculation jobs are provided by input nodes.
Necessary data nodes can be imported from files, managed by special plugins, or created from scratch specifically for running a calculation.
The process
buildercan be used to explore the inputs of a process, and set up a calculation step by step.The
verdiCLI allows you to follow the state of your calculations, get more information from the output nodes and check the in- and output files.